4. Flow objects
The cornerstone of EasyNMR is the flow objects that may be connected to give a specific functionality. There are more than 15 flow object types, and less than 10 flow transfer types. These numbers will increase as the functionality of EasyNMR is expanded. The description and function of flow objects are provided below.
4.1. Data objects
Data objects store and carry scientific data, tables, and molecules as described below.
4.1.1. Scientific data

EasyNMR stores scientific data in Data objects using the Core Scientific Data Model (CSDM). CSDM provides a lightweight, portable and self-contained model and file format for multi-dimensional scientific data. The CSDM object specifies the dimensions in the Dimensions pane and the dependent variables (data) residing on the grid specified by the dimensions are shown in the Dependent variables pane.
All scientific data that is uploaded to EasyNMR or dropped onto the flow window is automatically transformed to CSDM on load. See the In- and output section below for compatible file formats. If you have any wishes for support to other file formats, please reach out to us via email.
See our publication on the CSDM in PLOS ONE. Furthermore, there is a nice Python CSDM library, and CSDM is fully supported by the RMN, SIMPSON, and DMFit software packages.
In- and output
- Anchors:
- csdm:
The scientific data is output from the anchor in the CSDM format.
- File types:
- Bruker:
Bruker spectra are recognised as folders containing a fid or ser file and a pdata folder.
- Agilent:
Agilent spectra are recognised as folders with .fid extension and contain a file named fid.
- CSDM:
CSDM files are recognised as JSON files with file extension .csdf or .csdfe.
- ChemDraw:
ChemDraw XML files that contain predicted spectra are recognised by their extension .cdxml.
- SIMPSON:
SIMPSON files have the extension .spe or .fid.
- DPS:
File format of the Bruker MiniSpec TD-NMR, with extension .dps.
4.1.2. Table
The Table object stores tables of data. In the table pane, you can view and sort the content of a Table object. To create a table, either load an Excel or CSV file, or use the Calculator object.
The Import pane provides options for importing Excel and CSV files. For example, if your table has a title row, check Has title row; or if the elements of the table are separated by comma, write a comma in the Separator editbox.
In- and output
- Anchors:
- table:
The table input allows to present output from e.g. calculator object in tabular format. The output anchor emits the table to another object.
- File types:
- Excel:
Excel files have the extension .xls or .xlsx.
- CSV:
Files with comma-separated values have the extension .csv.
4.1.3. Text

The Text object allows adding rich text to your project. You can use this module to add descriptions or write notes. The text is visible in the Text pane of description. To modify the text, click on Control to expand the Control pane, and then click Edit. Now you can change the text, remember to save by clicking the disk icon. To exit the edit mode, click Done in the Control pane.
After you apply changes, the written text will appear in the Text pane and hides the editor.
In- and output
- File types:
- Text:
Text files have the extension .txt.
4.1.4. Molecule
The Molecule object loads molecules from PDB or CIF files. For CIF files, it is possible to expand the unit cell using the text input of the Molecule pane.
The Database pane offers the possibility to directly download an entry from the protein database.
In- and output
- Anchors:
- mol:
The mol anchor outputs the molecule for other objects.
- File types:
- PDB:
PDB files have the extension .pdb.
- CIF:
CIF files have the extension .cif.
- MOL:
ToDo: Files of the type MOL have extension .mol.
- XYZ:
ToDo: XYZ files have the extension .xyz.
4.2. Function objects
Function objects add functionality or calculations to NMR data as described below.
4.2.1. Calculator
The Calculator object takes any input through the in (data) and par anchors and makes this data available as input for a JavaScript operation. The JavaScript code should generate an output that is sent to the out anchor.
The Control pane shows the various inputs available along with their ID’s. Furthermore this pane allows configuration of the output type of the object. Optionally, choose to add a an empty output with the right format by clicking “Add empty output”.
The Calculation pane contains the JavaScript code for generating the output. Any input is available as part of the input variable with the ID as its key. By default, the Calculator object makes the output a copy of the first input, e.g.
output = input.WfeGwi;
If you had chosen to create an empty output, the default code would be
1var np = 128;
2output = {
3 version: "1.0",
4 dimensions: [{
5 type: "linear",
6 quantity_name: "frequency",
7 count: np,
8 complex_fft: true,
9 increment: "1 Hz",
10 label: "1H",
11 application: {
12 easy: {nucleus: "1H"}
13 }
14 }],
15 dependent_variables: [{
16 type: "internal",
17 numeric_type: "float32",
18 quantity_type: "scalar",
19 components: [
20 new Float32Array(np)
21 ]
22 }]
23}
In general, any JavaScript operation can be used, as long as the JavaScript code defines the variable output in the right format. If you use asynchronous code relying on JavaScript promises, you can also define the output to be such a promise that returns the output data on fulfill. For example, if we want to receive data from a web site, this could look like
1var csdm = input.WfeGwi;
2output = new Promise(fulfill => {
3 fetch('https://weather.data/somedata')
4 .then(res => res.json())
5 .then(data => {
6 // Assuming that the data object contains an array named arrayed_data
7 csdm.dependent_variables[0].components[0] = data.arrayed_data;
8 fulfill(csdm);
9 });
10});
In- and output
- Anchors:
- in:
A data input anchor to receive CSDM, table, or molecule data.
- par:
ToDo: The par anchor can receive and emit various parameters recognized and established by the parameter object.
- out:
The output from the calculation is forwarded to this anchor.
4.2.2. Simulation

The simulation object allows you to perform numerical simulations of NMR experiments through the EasyNMR interface. It interfaces to several simulation engines, most notably the widely used SIMPSON software. Since SIMPSON is a compiled program, such simulations need be run in a backend server provided by NMRBox. The simulation object seamlessly connects to the server and handles the output. Once the simulation is done, the resulting simulation is provided as a CSDM dataset at the CSDM anchor. In order to perform simulations you need to be authenticated at the eServer object (See eServer).
The SIMPSON input file (or input instructions for other simulation software) is shown in the Input pane of the simulation window with relevant syntax highlighting. The Control pane allows your to have control over how the simulations run. For example, you can choose a walltime, i.e. the maximum time for running simulations on the server and choose the output type. Press the Run button execute the simulation at the server. The status bar in the toolbar shows the progress of the simulation. A green checkmark appears on the Simulation module icon when the computations are finished. The Log pane displays the output from the simulation.
The Type pane allows to choose between different simulation engines. It defaults to SIMPSON but also allows to simulate deuterium dynamics using the software of F.H. Larsen in Annu. Rep. NMR 71, 103, 2010. Auxillary files (e.g. Tcl libraries for SIMPSON) required for the simulation may be added as zip files in the Data pane.
We have prepared a SIMPSON tutorial in EasyNMR here. This tutorial is published in Annu. Rep. NMR 100, 1, 2020 (open access preview).
In- and output
- Anchors:
- par:
ToDo: The par anchor can receive and emit various parameters recognized and established by the parameter object.
- data:
Anchor to receive generic data that will be transmitted to the server as a file with the name is listed in the Data pane.
- spins:
ToDo: Receive a spin system from e.g. a SIMMOL object.
- csdm:
The resulting spectrum of the simulation is output in the CSDM format.
- File types:
- SIMPSON:
SIMPSON input files have the extension .in and are opened using this object.
4.2.3. Model

The Model object provides a comprehensive, versatile, and yet simple way to peform numerical simulations of your scientific data. Currently, the Model object focuses on modelling NMR data, but the interface is by no means limited to that.
The scientific data to fit should be connected to the csdm anchor. Once one or more datasets are connected, they appear in the Datasets pane, where the dropbox allows you to select to include the dataset in the simulation, to ignore it, or to allow to add it as a line in a simulation (see later). The feature to enable several experimental datasets in the same modelling setup allows to determine nuclear spin interaction parameters from e.g. multifield studies or experiments with different spin rates, etc.
The modelling of data is controlled by a number of different parameters. The Model object organises parameters in three groups
Global parameters. These can be defined and will be available in all simulations.
Line parameters: These parameters are associated with the individual lines required to fit the dataset.
Dataset parameters: A set of parameters that are associated with the individual datasets, e.g. spectral width, spin rate, Larmor frequency.
Datasets selected to include in the simulation appear in the top line of the Lines pane, and you may press the individual datasets to display the parameters for the specific dataset. Below the dataset there are three sections:
The line parameters and the possibility to add new lines or delete existing lines of the model.
The dataset parameters (see above)
ToDo: Definition of the data stored in the output dataset. This feature is not yet implemented, so the resulting dataset just contains two dependent variables: The input (experiment) and the simulation.
When adding lines to the modelling of a dataset, select between the different line models. Easy line model comes with a default set of parameters, which may be modified by pushing the “Edit” button. Remember that quantities are numbers and units! For each parameter, it is possible to select if the parameter should be optimized or fixed during an optimization. If there are global parameters of matching type (i.e. same quantity name etc), it is also possible to bind a line parameter to one of the global parameters. This is relevant if two datasets should be fitted with the same set of parameters, e.g. same quadrupole coupling constant.
Global parameters are defined and listed in the Global parameters pane. To add, remove, or change a global parameter, push the “Edit” button in the top right corner of the Global parameters pane. To add a parameter, give it a name, a value, and select the type. If you select a quantity, you also need to specify the quantity name. Hit “Add” to add the parameter. Once the global parameter is added, you will notice that all Dataset and Line parameters of same type now have more control options: Optimize, Fix, or bind to the global parameter.
The line types are mostly self explained, but you will find the custom line model under the menu . This allows you to generate JavaScript code that returns the simulated data. The global and dataset parameters are available as par.dataset.spin_rate, par.global.cq etc, and the frequencies (the x-axis) is available as the variable x (which is an array of n arrays for an nD experiment). The JavaScript code should return an array of same length as the grid size with the simulated values.
In- and output
- Anchors:
- csdm:
The experimental data to be modelled are sent to the object through this anchor.
- par:
ToDo: The par anchor can receive and emit various parameters recognized and established by the parameter object. These parameters are defined as global parameters.
- out:
The resulting spectra of the simulation is output in the CSDM format.
4.2.4. eServer

The eServer object (short for EasyNMR Server) allows signing in to the backend server. This is in general not required to use EasyNMR, except for a few functionalities requiring access to the backend server. These functionalities include
Running SIMPSON simulations using the Simulation object
Performing automatic peak picking in the Plot object using the NMRGlue peak picker
Without eServer access, these functions will not be available in EasyNMR.
The eServer uses ORCID for authentication. When pushing the Login button, you are directed to ORCID. EasyNMR only asks for permission to authenticate, not to modify anything in your ORCID account, so it should be safe to authorize EasyNMR to use ORCID to authenticate. After successfully logging in, you see the server you are connected to (e.g. easynmr.nmrbox.org), its status, your name, your ORCID identification, and the capabilities available with the server. You can log out from the server by pressing the Log out button.
The Lost files pane displays any files from simulations not transferred to your EasyNMR workspace, e.g. if your computer lost its connection to the server during a simulation. Such files may either be loaded or removed.
The Share workflow pane allows you to upload the current EasyNMR workflow to the eServer and obtain a unique link to the workflow for easy sharing.
The Administration pane is only for EasyNMR system administrators.
4.2.5. SIMMOL

The SIMMOL object has a two-fold function. To visualise molecules and nuclear spin interactions and to establish a spin system in a molecule.
The Molecules pane shows which molecules are attached to the object, and for each molecule it gives a ID and other information. ToDo: Right now the visualisation only shows the first molecule, but the SIMMOL commands work for all molecules.
The Control pane allows to switch on and off various visual effects and to reset the view. The Scene pane displays the molecule and lets the user interact with the molecule. It is possible to rotate, zoom and pan. Furthermore, it is possible to select individual atoms by clicking on them in the Scene pane. Once an atom is selected, it changes color and information about the atom is displayed in the Result pane. If the Easy layout is saved once the molecule has been rotated or zoomed, the new scene is saved. Use the ‘Reset view’ button in the Control pane to reset it. The molecule visualisation is provided by ChemDoodle Web.
The SIMMOL pane is a text editor that accepts JavaScript input. The molecules are available through the object mol that stores each molecule by its ID keyword, thus the instruction
var m = mol.hUqPDs;
defines the variable m as the array of molecules with the ID hUqPDs. A molecule, e.g. m[0], is represented as an object with arrays of atoms and bonds and other information used by the ChemDoodle visualiser. The atom object is relevant to know, as its keywords may be used for selection of atoms. The following atom is from loading PDB ID 1UBQ and showing atom 378 (i.e. m[0].atoms[378]):
1{
2 "label": "N",
3 "x": 32.478,
4 "y": 30.917,
5 "z": 22.269,
6 "info": {
7 "id": 321,
8 "resname": "ARG",
9 "resid": 42,
10 "chain": "A",
11 "name": "N"
12 },
13 "hetatm": false,
14 "isotope": {
15 "name": "14N",
16 ...
17 },
18 ...
19}
Selection of atoms from a molecule are done through normal JavaScript functions like filter. As an example, the following command would select all amide nitrogen atoms
var nitrogens = m[0].atoms.filter(a => a.info.name == "N");
Selections can be combined in one command (line 1 below) or nested (lines 2-4), so to select the amide nitrogen of residue 42, the following two commands would both provide the right result
1var n42 = m[0].atoms.filter(a => a.info.name == "N" && a.info.resid == 42);
2var n42 = m[0].atoms
3 .filter(a => a.info.name == "N")
4 .filter(a => a.info.resid == 42);
Once the relevant list of atoms is selected, different SIMMOL commands may be applied to the selection. See the table below for a list of SIMMOL commands:
matoms |
matoms [atom list] ?{color: [r g b], radius: r}? Highlights the atoms provided as input argument. These atoms are visible as SIMMOL annotations in the Scene pane. The options specification is optional, and each of the keywords can be left out |
mbonds |
mbonds [atom list 1] [atom list 2] ?[r g b]? Creates and displays bonds between all atoms in the two atom lists. The color of the bonds may optionally be specified |
mplane |
plane [atom list] ?[r g b]? The command assumes that the atom list contains a list of amide nitrogen atoms. It establishes the peptide plane for each amide nitrogen and visualises it |
mshift mquadrupole |
mshift/mquadrupole [atom list] ?{angles [α β γ], coordsys: [xx xy xz yx yy yz zx zy zz], color: [r g b], cutcolor, [r g b]}? For atoms connected to a peptide plane, a standard orientation is used (see the original SIMMOL paper). For other atoms, the angles and coordinate system need be specified. In all cases, the color of the ellipsoid and the color of its cut may be defined |
mdistance |
mdistance [atom list 1] [atom list 2] Calculates and visualises the distance between atoms in list 1 and 2 |
mdipole |
mdipole [atom list 1] [atom list 2] ?[r g b]? Calculates and visualises the dipole-dipole coupling between atoms in list 1 and 2. It uses the information about the isotope of each atom |
mcoordsys |
mcoordsys <atom 1> <atom 2> <atom 3> Returns a coordinate system (to use with mshift and mquadrupole) based on the three atoms specified. The coordinate system is centered at atom 2. X is along the atom 1 ↔︎ atom 2 bond, Z is perpendicular to the plane spanned by the three atoms, and Y is perpendicular to X and Z. |
maddHydrogen |
maddHydrogen <atom> ?type? Adds missing hydrogen atoms to the specified atom (carbon or nitrogen). The type indicates if the atom is “sp”, “sp2”, or “sp3” hybridized. It defaults to sp3. |
mdist2dip |
mdist2dip <isotope 1> <isotope 2> <distance> Calculates the dipole-dipole coupling for two isotopes separated by a certain distance |
mdip2dist |
mdip2dist <isotope 1> <isotope 2> <dipolar coupling> Calculates the distance two isotopes having a certain dipole-dipole coupling |
msphere |
msphere [x y z] radius [r g b] Displays a sphere in the Scene pane with at a given point in space with a given radius and color |
mtriangle |
triangle p1 p2 p3 [r g b] Creates and displays a triangle between the three points (p1 = [x1 x2 x3] etc) with a given color |
moctahedron |
moctahedron [p1 p2 p3 p4 p5 p6] [r g b] Creates and displays an octahedron between the six points (p1 = [x1 x2 x3] etc) with a given color |
ToDo: The spin system output is not yet implemented in the SIMMOL object.
In- and output
- Anchors:
- mol:
The object receives a molecule from another object through this anchor.
- spins:
ToDo: The par anchor can receive and emit various parameters recognized and established by the parameter object. These parameters are defined as global parameters.
4.2.6. Trace
The function of the Trace object is to extract cross sections of multidimensional scientific data. Currently, it only supports 2D input and 1D output, but in principle, you could also imagine taking a 2D cross section of a 5D dataset. That will come later.
The Trace/Projection pane allows selection of the desired type of trace. You can select if the trace should be horizontal or vertical, and if it should be just a trace or an integral region. Regions/traces can be specified either as points (e.g. “1 pt”), quantities (e.g. “1200 Hz”), or ppm if available (e.g. “12.01 ppm”). If there are two dependent variables in the dataset, there will also be the option to obtain the ratio between the two datasets - this is very relevant for processing of REDOR NMR experiments.
The output will be a 1D dataset, where the dimension will be a copy of the first dimension of the 2D input dataset for horizontal traces and the second dimension for vertical traces.
In- and output
- Anchors:
- in:
The 2D spectrum from which a trace/integral should be extracted is sent to this anchor.
- out:
The resulting 1D trace/integral of the simulation is output in the CSDM format.
4.2.7. Plot
The plot object can plot scientific data from single and multiple sources. Different datasets can be overlaid for comparison. The Plot layout pane allows to select the plot type as 2D or 3D (contour or heatmap) and below displays a list of datasets connected to the plot object compatible with the selected dimensionality.
Using the checkbox in front of each dataset, these may be selected or deselected for plotting. Note that dependent on the axis settings, some datasets may not be selectable, e.g. if they have different quantities. Once a dataset is selected, its plot type may be configured (2D: lines, markers or lines+markers, 3D: heatmap or contour) and the plot may be configured.
To configure the axis settings, click Configure x/y axis to display configuration options for the axis. Most options are self explained. The axis type may be switched between Index (all datapoints are numbered sequentially), Quantity, or Dimensionless (if available).
The Plot pane shows the interactive plot with the possibility of zooming and panning. The plot may be downloaded either in PNG or SVG (vector based) formats. It is also possible to move the legend between the four corners or to toggle it off.
For NMR frequency-domain data it is possible to activate peak picking and integration. When doing peak picking and integration, the peak/integration information becomes available in the Annotations pane. If you have authenticated to the eServer, automatic peak picking using NMRGlue will be available in the Control pane. These annotations may be saved with the CSDM object.
In- and output
- Anchors:
- csdm:
The scientific data to plot are submitted to the object through this anchor.
4.3. Auxiliary objects
4.3.1. File

The File object allows you to download a copy of NMR data in the CSDM format or to save a table object as an Excel file.
To save data as a file click on the file icon in the Toolbar to create a File object. Drag the File object to the desired place in the project view. Only certain components can be connected to the File object. Compatible objects include: Scientific Data (CSDM), Table, and SIMPSON.
To connect the File object to a compatible component, click on the connect icon and then on the anchor of the newly created File object. Anchors of other compatible components will blink. Click on the component of your choice. Components already connected to the File component will not blink.
In- and output
- Anchors:
- data:
Data of any kind (see above for limitations) will be available for download.
4.3.2. Configuration

The Configuration object displays and edits default preferences used by EasyNMR. For example, when a new Scientific Data is added to the Workflow, a new Plot object is added and connects to the new data object.
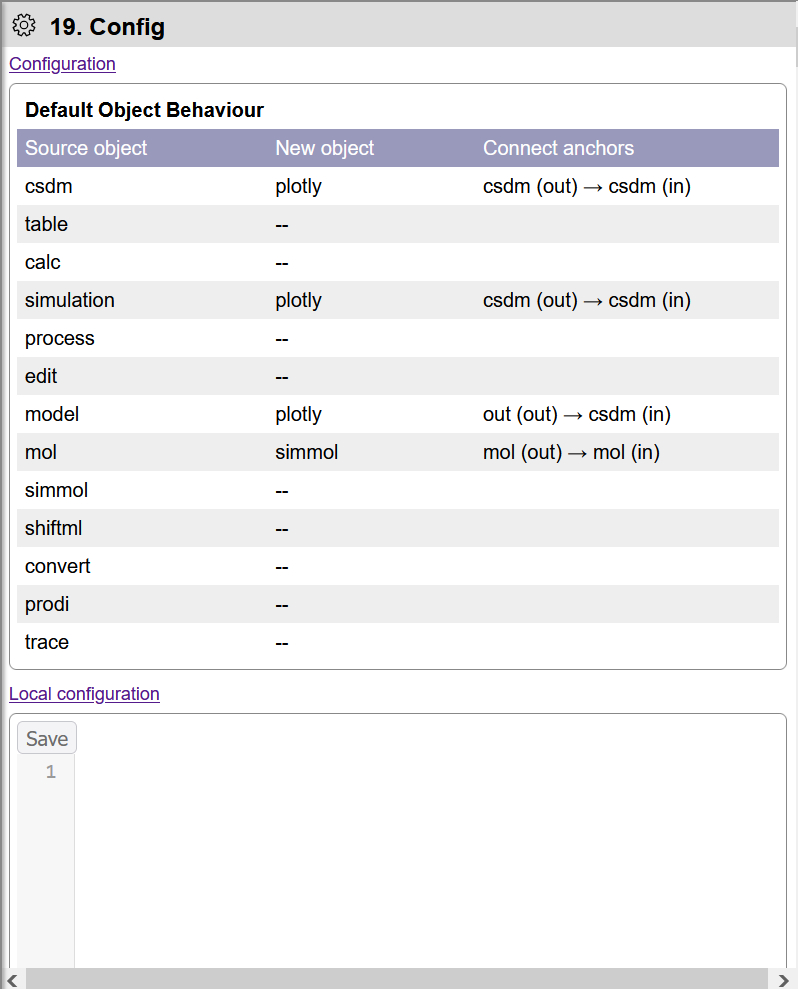
Default object behavior in the Configuration object.
4.4. Transient objects
Transient objects are part of the Workflow that are produced if required, but will not be a permanent part of it, for example when exporting it.
4.4.1. Folder

Drag and dropping a folder containing several NMR data sets creates a Folder object from which you can extract individual 1D/2D spectra as Scientific Data (CSDM) objects. The Object Properties of Folder shows all NMR spectra contained and text files in the folder. Press on Extract to create a data object from a specific processed experiment.
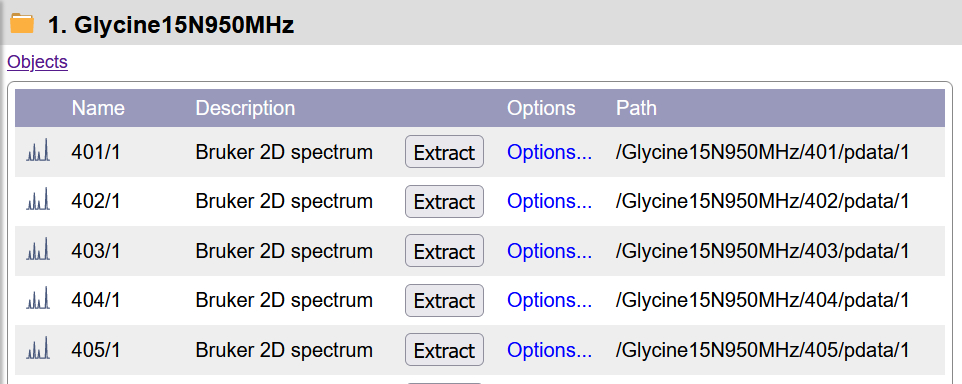
List of NMR experiments in a Folder.
4.4.2. Archive

Drag and dropping an archive (for example, a zipped file) containing several NMR data sets creates an Archive object. In the Object Properties of an archive object, you can see a list of experiments and extract the ones you are interested in as Scientific Data objects.
Note
Always click on the connect icon to deactivate the connect tool after connecting the desired objects.
Tip
You can change the position of the anchor the left and right of an icon by clicking on the Anchors pane of File Object Properties.
Tip
For each object, you can see relevant help by clicking on the Documentation pane to expand it in the Object Properties window.